Quality assessment description
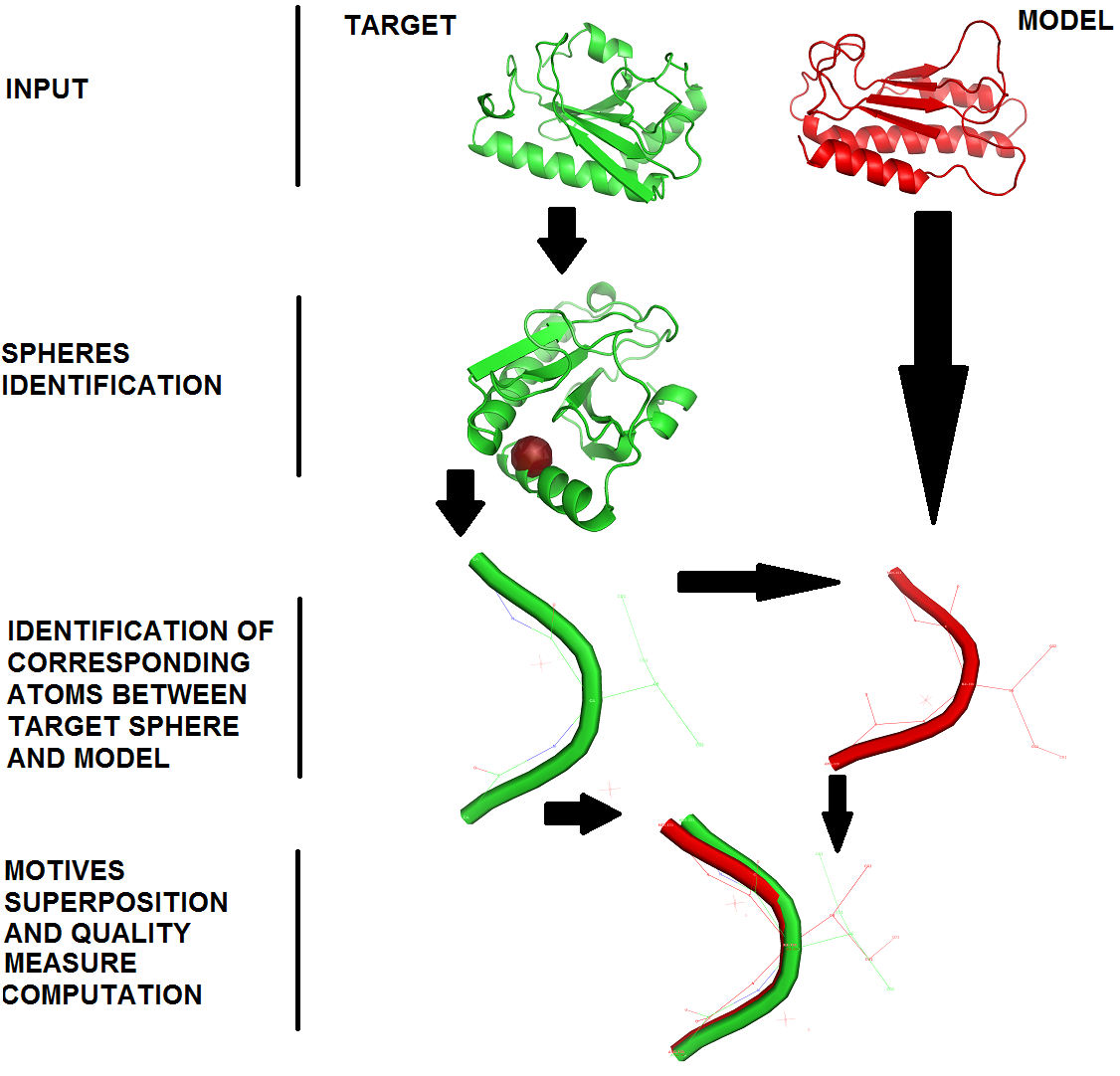
Figure 1. Quality assessment routine diagram.
As input a user should set some basic confi guration parameters as following: the reference structure (marked in green); a structure of the predicted model (marked in red); the selected atom type treat as a sphere center, sphere radiuses vector which describes interesting accuracy levels; calculation mode which decides whether all or only selected atoms from each amino-acid of analyzed molecules will be analyzed. Generaly, structural conformity evaluation between a predicted protein model and its reference structure is conducted by comparison of corresponding atoms sets. For every amino-acid of the reference structure the sphere is built. The atoms set of the reference structure is localized inside a sphere which is built on central atom de fined by user (e.g. 'CA') with a strictly identified radius. As a result of the sphere building process we obtain a set of atoms that are included in a particular sphere. Next, for every sphere built on the reference structure, a corresponding set of atoms in the predicted model is identifi ed. Finally, the corresponding structural motives are superimposed, and the quality measure (e.g. RMSD) of arisen structural alignment is computed. The sphere radius value uniquely identi fies the analysis accuracy level (e.g. lower sphere radius represents local perspective; otherwise increasing sphere radius is trying to achieve global perspective).
What you can do with the SphereGrinder tool?
The SphereGrinder allows the user to evaluate the agreement between a predicted model of an protein molecule and its reference structure.The proposed software was designed according to module-based architecture and mainly consists of a computational module and a visualization module. The computational module is used to compute the values of RMSD measure between the particular model and the reference structure based on sets of atoms located inside spheres identifi ed by user taking into account sphere radiuses vector in discrete space. For each protein model, de fined by user, the comparison between it and its reference structure is performed separately. Before starting the computation one can set or change the set of analyzed models, their reference structure and the vector of sphere radiuses as well as the central atom which will be used as a sphere center. The user can also decide whether all or only selected atoms from each amino-acid of analyzed molecule will be considered. This module allows also for inconsistencies identifi cation from input PDB files which are reported to user in a Log view. Every inconsistent atom record is skipped during computational analysis. The visualization module consists of five types of plots which represent di fferent views and levels of quality accuracy. Our software allows also the user to visualize the results from the entire accuracy levels vector in one plot simultaneously.
System requirements
- » Java Platform, Standard Edition 6.0 http://java.com
- » Java 3D 1.5.2 https://java3d.java.net/binary-builds.html
- » OpenGL http://www.opengl.org/wiki/Getting_Started
When you have any problems make sure that you have installed Oracle Java SDK 1.6 and Java3D 1.5.2. When you have installed modern Java SDK (7 or 8) use following guide HowToRunSphereGrinder.pdf.
How you can cite SphereGrinder?
Piotr Lukasiak, Maciej Antczak, Tomasz Ratajczak, Marta Szachniuk and Jacek Blazewicz "Quality assessment methodologies in analysis of structural models", Proceedings of the 25th European Conference on Operational Research, pp. 80, 2012